PCR Anreicherung der Zielregionen und Illumina Hochdurchsatzsequenzierung (Next Generation Sequencing) mit anschließender bioinformatischer Auswertung.
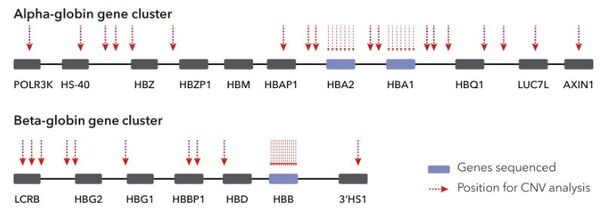
Bei der Interpretation der Testergebnisse ist es wichtig zu beachten, dass die Schwere der Thalassämie von der Anzahl und Art der betroffenen Hämoglobin-Gene abhängt. Thalassämien werden vererbt, und wenn beide Elternteile Träger bestimmter Thalassämieformen sind, besteht ein erhöhtes Risiko für die Geburt eines Kindes mit schwerer Thalassämie. Mögliche Formen der Ausprägung sind:
Alpha-Thalassämie:
Beta-Thalassämie: